GSK-3 (glycogen synthase kinase) is a multifunctional serine/threonine kinase that is active all the time in all cells, but particularly highly in the brain. GSK-3 was originally named for its ability to phosphorylate and inactivate glycogen synthase and regulate glucose metabolism. As genes and proteins are discovered, they are often ascribed names based on function—their function at the time of discovery. GSK-3 turned out to be a multifunctional enzyme, able to phosphorylate many proteins. Phosphorylation of the protein acts as a molecular switch, turning the activity on or off. GSK-3 has many phosphorylation targets. It should therefore not be surprising that GSK-3 has both pro- and antiapoptotic roles.
GSK-3 is presently known to be a key regulator of a wide range of cellular functions. GSK-3 regulates numerous cellular processes through a number of signaling pathways important for cell proliferation, stem cell renewal, apoptosis and development. Because of these diverse roles, dysregulated GSK-3 has been implicated in several diseases including neurodegenerative diseases (Alzheimer’s and Parkinson’s disease), stroke, bipolar disorder, type-2 diabetes, inflammation and cancer.
Phosphorylation of a protein by GSK-3 usually inhibits the activity of its downstream target. Thus, signals that inhibit GSK-3 often cause activation of its diverse array of target proteins. Regulation of GSK-3 is important for normal development, regulation of metabolism, neuronal growth and differentiation and modulation of cell death. GSK-3 is encoded by two genes, GSK-3α and GSK-3β, which are structurally similar, but functionally non-identical. There is a homologous serine on both these isozymes; Ser9 on GSK-3α and Ser-21 on GSK-3β. Phosphorylation of this serine by an upstream kinase, such as Akt, inactivates GSK-3 and prevents it in turn from phosphorylating its own downstream targets.
Glycogen synthase kinase 3: a key regulator of cellular fate.
GSK3 as a Sensor Determining Cell Fate in the Brain.
Emerging roles of glycogen synthase kinase 3 in the treatment of brain tumors.
What Are the bona fide GSK3 Substrates?
In cancer, GSK-3 modulates the response of the cell death machinery to stress stimuli, including chemotherapeutics. Mitochondria are at the heart of the integration between survival and death signals; therefore, modulation of the mitochondrial functions carried out by GSK-3 is profoundly involved in the apoptosis escape capabilities. In cancer tissues, the high glycolytic activity requires an up-regulation of the key glycolytic enzymes including hexokinase. GSK-3 also regulates tumor cell survival by controlling mitochondrial binding of hexokinase, particularly hexokinase type II, which is highly expressed on the outer mitochondrial membrane of most cancer cells. Hexokinase (type I and II) bind to a protein in the mitochondrial membrane called VDAC. VDAC is a dynamic regulator of global mitochondrial function.
Hexokinase initiates the process of intracellular glucose utilization and it contributes to the Warburg effect, the aerobic glycolytic metabolism of cancer cells. Hexokinase (type I and II), the rate limiting enzymes in glycolysis, controls cell survival by promoting metabolism and/or inhibiting apoptosis. Inhibition of glycolysis severely depletes ATP in cancer cells and leads to massive cell death. It was found that release of Hexokinase II from mitochondria induces apoptosis in hepatocellular carcinoma and glioma cells. When hexokinase is detached from mitochondria, the mitochondrial membrane undergoes conductibity changes which increases its permeability, promotes the release of cytochrome C into the cytosol and activates caspase-3. Caspase-3 activation leads to apoptotic cell death.
GSK-3 and mitochondria in cancer cells.
The modulation of inter-organelle cross-talk to control apoptosis.
Regulation of hexokinase binding to VDAC.
Hexokinase binding to mitochondria: a basis for proliferative energy metabolism.
Apoptosis-inducing antitumor efficacy of hexokinase II inhibitor in hepatocellular carcinoma.
Elemental lithium, a drug widely used to treat bipolar mood disorder, is a well established inhibitor of GSK-3 activity. Lithium binds to GSK-3 to directly inhibit its activity. In an alternate, indirect way, lithium increases the inhibitory phosphorylation of a critical serine residue in GSK-3, causing its inactivation. This makes lithium a powerful inducer of both autophagy and apoptosis by multiple biochemical pathways. Another study shows that lithium also detaches hexokinase from the mitochondrial membrane.
Emerging roles of glycogen synthase kinase 3 in the treatment of brain tumors.
Inhibition of GSK3 by lithium, from single molecules to signaling networks.
Lithium detaches hexokinase from mitochondria and inhibits proliferation of B16 melanoma cells.
GSK-3 also plays a fundamental role in the initiation of inflammation. Inflammation is a critical component of solid tumor progression. Many cancers arise from sites of infection, chronic irritation, and inflammation. GSK-3 promotes the production of inflammatory molecules and cell migration, which together make GSK-3 a powerful regulator of inflammation, while GSK-3 inhibition provides protection from inflammatory conditions.
NF-kB, a major transcription factor, provides a critical mechanistic link between inflammation and cancer. GSK-3 promotes NF-kB induced gene transcription and enhances inflammation primarily by activating NF-kB activity in the nucleus. GSK-3β has been shown to participate in NF-kB mediated cell survival in pancreatic cancer and acute lymphocyte leukemia (ALL), thus behaving as a cancer promoter. Thus, inhibition of GSK-3β results in inhibition of the NF-kB pathway and reduction of NF-kB-mediated transcription. GSK-3 inhibitor lithium exhibits anti-inflammatory and anti-tumor effects. Lithium modulates pro- versus anti-inflammatory cytokines and pro- versus anti-apoptotic gene expression. Lithium can be considered an anti-inflammatory agent.
Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics.
TNF-α (tumor necrosis factor-alpha) is a cytokine with antitumorigenic property. The interaction of TNF-α with TNF receptor (TNFR-1, TNFR-2) activates several signal transduction pathways, leading to the diverse functions of TNF-α. TNF-α activates both apoptotic pathways along with survival and proliferation pathways via TNFR-1. Low dose, chronic TNF-α production by tumor cells promotes tumor growth and metastasis.
Lithium promotes TNF-α production, thereby stimulating apoptosis in cancer cells. Lithium-induced cell death is largely mediated by the release of TNF-α. Furthermore, when GSK-3 is inhibited by lithium, apoptosis is promoted by low levels of TNF. That is, lithium enhances the sensitivity of cancer cells to TNF. It was found that the inhibition of GSK-3 has a sensitizing effect on TRAIL-induced apoptosis both in prostate and pancreatic cancer. In addition, the inhibition of GSK-3 promotes the TRAIL (TNF-related apoptosis-inducing ligand) cellular death pathway.
Unfortunately, TNF-α also stimulates the activation of NF-kB which blocks its ability to induce apoptosis in cancer and leukemia cells. Another study claims that GSK-3 inhibition does indeed reduce NF-κB activity but does not result in TNF-mediated apoptosis, potentially due to the activation of pro-survival genes through Wnt signaling. Wnt is secreted by cells. Wnt proteins are ligands, which means they can attach (bind) to other proteins called receptors. The free Wnt molecule binds it’s receptor on the cell membrane and activates a survival/growth pathway. Wnt activates a protein called beta-catenin, and this is thought to contribute to the progression of many cancers. GSK-3 has dual functions in the regulation of cell survival, where it can either activate or inhibit apoptosis, further complicating its involvement in cancer.
TNF-alpha in cancer treatment: molecular insights, antitumor effects, and clinical utility.
Glycogen synthase kinase-3 inhibition sensitizes pancreatic cancer cells to TRAIL-induced apoptosis.
LiCl induces TNF-α and FasL production, thereby stimulating apoptosis in cancer cells.
Wnt pathway activity confers chemoresistance to cancer stem-like cells in a neuroblastoma cell line.
A Wnt-ow of opportunity: targeting the Wnt/beta-catenin pathway in breast cancer.
APC shuttling to the membrane, nucleus and beyond.
Cancer is caused by mutations in critical genes, such as p53. Mutations in many cancer-related genes can disrupt apoptosis, resulting in resistance to common anticancer therapies. In the absence of apoptosis, autophagic cell death can be an alternative form of cell death by excessive self-digestion. If autophagy is prolonged in cancer cells, they can self destruct. Therefore, autophagic cell death can be considered as a backup cell death mechanism when apoptotic cell death mechanisms fail. GSK-3β signaling is a key mechanism in regulating autophagy activation. NF-kB inhibits the induction of autophagy by both TNF and TRAIL. Lithium inhibits the activation of GSK-3, thereby completely inhibiting the activation, nuclear transport and DNA binding of NF-kB.
Understanding autophagy in cell death control.
GSK-3beta promotes cell survival by modulating Bif-1-dependent autophagy and cell death.
NF-kappaB activation represses tumor necrosis factor-alpha-induced autophagy.
Angiogenesis (blood vessel growth) is critical for invasive tumor growth and metastasis and constitutes an important point in the control of cancer progression. GSK-3β/beta-catenin axis promotes angiogenesis through activation of VEGF (vascular endothelial growth factor) signaling in endothelial cells. Beta-catenin is an important modulator of angiogenesis. VEGF plays a critical role in angiogenesis due to its specific ability to promote the proliferation and migration of endothelial cells. NF-kB is also involved in the upregulation of VEGF and mediates angiogenesis.
The rate of angiogenesis is related to the rate of glycolysis. To fulfill tumor cell needs, the glycolytic switch is associated with elevated glucose uptake and lactic acid release. Lactic acid also induces angiogenesis. Lithium suppresses angiogenesis through inhibition of GSK-3β signaling pathway. However, another study shows that lithium promotes VEGF expression through PI3-K/GSK-3β-dependent and -independent pathways in brain endothelium and astrocytes, respectively. This growth factor (VEGF) signaling mechanism may contribute to lithium’s reported ability to promote neurovascular remodeling after stroke.
Lithium upregulates vascular endothelial growth factor in brain endothelial cells and astrocytes.
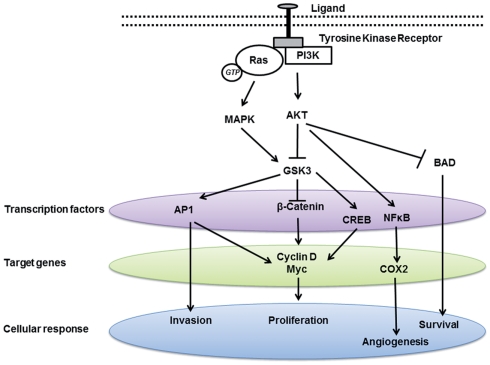
GSK-3 is upregulated in many types of tumor, including prostate, colon, stomach, pancreas and liver cancer. GSK-3 is required both for hormone-dependent and hormone-independent prostate cancer growth. However, some studies suggest a conflicting role of GSK-3β in various cancers, either as a tumor suppressor or tumor promoter. For example, GSK-3β has been shown to inhibit androgen receptor-stimulated cell growth in prostate cancer, thus acting as a tumor suppressor. In contrast, other study showed that suppressing GSK-3β activity reduced prostate cancer cell growth. GSK-3β is also highly expressed in colon cancer, prostate cancer, etc. and has been shown to participate in NF-κB mediated cell survival in pancreatic cancer, thus behaving as a tumor promoter. It is clear that different signaling pathways regulate GSK-3 activity by different mechanisms.
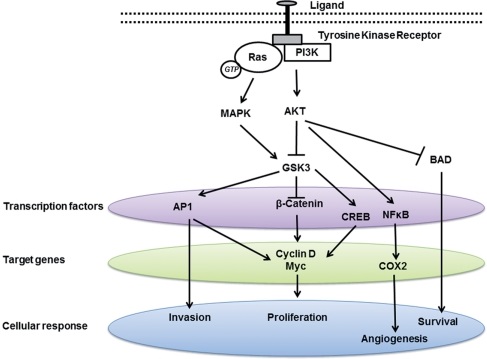
Inhibition of GSK-3 beta activity attenuates proliferation of human colon cancer cells in rodents.
GSK-3β: A Bifunctional Role in Cell Death Pathways.
Distinct expression and activity of GSK-3α and GSK-3β in prostate cancer.
Suppression of glycogen synthase kinase 3 activity reduces tumor growth of prostate cancer in vivo.
Aberrant glycogen synthase kinase 3β in the development of pancreatic cancer.
Glycogen synthase kinase-3β, NF-κB signaling, and tumorigenesis of human osteosarcoma.
Glycogen synthase kinase-3beta positively regulates the proliferation of human ovarian cancer cells.
Regulation of glycogen synthase kinase-3 beta (GSK-3β) by the Akt pathway in gliomas.
Glycogen synthase kinase–3β inhibitors suppress leukemia cell growth.
GSK-3 inhibitor lithium modulates cancer cell growth, apoptosis, gene expression and cytokine production in many different types of cancer and leukemia. Lithium specifically inhibits Hedgehog signaling pathway through the inhibition of GSK-3β. GSK-3β also plays a significant role in Hedgehog signaling. The Hedgehog signaling pathway plays a critical role in the initiation and development of cancer. Blocking Hedgehog pathways induces cancer stem cell death. In a normal person, the Hedgehog signaling pathway is under inhibition and gets activated upon the binding of Hedgehog ligand to a transmembrane receptor called Patched (PTCH1).
The combined synergistic use of lithium and a few other natural agents can suppress tumor growth through distruption of glycolysis and angiogenesis and induce apoptosis and autophagy. Lithium carbonate and lithium chloride are prescription drugs, but lithium orotate can be purchased as a supplement. Lithium orotate is a salt of orotic acid and lithium. There are no systematic reviews of the efficiency of lithium orotate for any condition. In 1979, it was found that lithium orotate was more dangerous to the kidneys than lithium carbonate. 300 mgs of lithium carbonate contains 56.4 mgs of elemental lithium. The normal oral dose of lithium carbonate for the treatment of bipolar disorder starts at 600-900 mgs a day 2-3 divided doses. These therapeutic doses inhibit GSK-3 activity. The synergistic use of lithium and a few Herbalzym products could have a profound affect on the cellular functions of tumor suppressor genes and the promotion of cancer cell death. But, don’t take lithium unless it’s under the supervision of an occupational therapist. Because toxicity can occur at levels >1.5 mEq/L, lithium levels must be carefully monitored especially during the first few weeks and lithium dosage adjusted as necessary.
Hedgehog signaling pathway and cancer therapeutics: progress to date.
Hedgehog signaling pathway: the must, the maybe and the unknown.
Hypoxia triggers hedgehog-mediated tumor-stromal interactions in pancreatic cancer.


{kind=link}