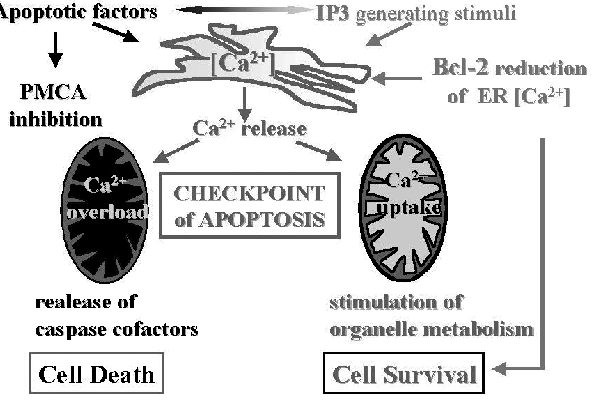
It has long been known that calcium ions Ca(2+) signals govern a host of vital cell functions and so are necessary for cell survival. However, more recently it has become clear that cellular calcium ions Ca(2+) overload, or perturbation of intracellular Ca(2+) compartmentalization, can cause cytotoxicity and trigger either apoptotic or necrotic cell death.
Many cellular processes require proper cooperation between the plasma membrane, the nucleus and subcellular vesicular/tubular networks such as the ER (endoplasmic reticulum) and mitochondria. It has recently become clear that such contacts are crucial for the synthesis and intracellular transport of phospholipids as well as for intracellular Ca(2+) homeostasis, controlling fundamental processes like motility and contraction, secretion, cell growth, proliferation and apoptosis.
Altered metabolism is a feature of many diseases, as well as aging. The definition of this essential mechanism for regulating cell energy will have implications for a wide variety of physiological processes and diseases such as cancer. Most healthy cells in the body rely on a complicated process called cellular-respiration (oxidative-phosphorylation) to produce the fuel ATP. Cellular-respiration (oxidative-phosphorylation) occurs in the mitochondria, where a series of enzymes catalyze the transfer of electrons to molecular oxygen and the generation of energy-storing ATP. A fundamental control system regulating ATP is an ongoing shuttling of calcium to the mitochondria from another cell component called the ER (endoplasmic reticulum).
The ER (endoplasmic reticulum) is the major reservoir of calcium in cells. The stored calcium is released to adjacent mitochondria through a calcium ion channel called the IP3 receptor. Recent study shows that this calcium release occurs at a low level all the time. When the researchers interfered with the calcium release using genetic or pharmacological methods, the mitochondria were unable to produce enough ATP to meet the needs of the cell. This indicates that mitochondria rely on the ongoing calcium transfer to make enough ATP to support normal cell metabolism. In the absence of this transfer, the mitochondria fail to make enough ATP, which triggers an extreme cell survival process called autophagy, or self eating.
Autophagy is important for clearing aggregated proteins from cells, for example in neurodegenerative diseases, and it plays a role in cancer. The IP3 receptor plays important roles in the regulation of programmed cell death, a process that is subverted in many cancers, and in neurodegenerative diseases. Calcium release from the IP3 receptor may be at the nexus of cancer and the role of cell metabolism gone awry in these broad disease classes. Uptake of Ca(2+) by mitochondria serves as a regulator of a number of important cellular functions, including energy metabolism, cytoplasmic Ca(2+) signals, and apoptosis. It is now known that Ca(2+)-dependent processes are interwoven with the mainstream apoptosis executioners-The caspases- and interfering with the sequestration of Ca(2+) into the intracellular pools, that is , the ER (endoplasmic reticulum), can be sufficient to trigger apoptosis as part of stress response.
The role of mitochondria in apoptosis.
Regulation of cell death: the calcium-apoptosis link.
Ca2+ signaling, mitochondria and cell death.
Calcium orchestrates apoptosis.
Mitochondrial calcium signalling: message of life and death.
SR/ER-mitochondrial local communication: calcium and ROS.
High- and low-calcium-dependent mechanisms of mitochondrial calcium signalling.
Calcium signaling: double duty for calcium at the mitochondrial uniporter.
Apoptosis and calcium: new roles for cytochrome c and inositol 1,4,5-trisphosphate.
The induction of apoptosis is dependent on the availability of proapoptotic proteins Bax and Bak, which are essential effectors of the mitochondrial outer membrane permeabilization (MOMP) and subsequent caspase activation. Tumor cells deficient in the Bak and Bax are resistant to chemotherapeutic drugs. Recent study shows that murine embryonic fibroblasts deficient for both Bak and Bax are, however, efficiently killed by a inhibitor of intracellular calcium pumps that induces ER stress by depleting ER Ca(2+) stores. In the presence of Bak and Bax, inhibitor of calcium pumps eliminates cells by release of mitochondrial cytochrome c and subsequent caspase activation, which leads to the proteolytic inactivation of the molecular necrosis switch PARP-1 and results in apoptosis.
Caspase-3 and -9 are activated in human myeloid HL-60 cells by calcium signal.
Various oncoproteins and tumor suppressor proteins physically localize to the mitochondria in cancer cells where they directly regulate malignant mitochondrial programs, including apoptosis. Redox status of mitochondria is important in combating oxidative stress and maintaining membrane permeability. Mitochondrial events involved in apoptosis and necrotic cell death, such as activation of Bcl-2 family proteins, formation of permeability transition pore, release of cytochrome c and apoptosis inducing factors, activation of caspase cascade, and ultimate cell death is the focus of attention in unraveling stress responses.
Oxidant stress led to a phenotypic shift in Ca(2+) mobilization from an oscillatory to a sustained elevated pattern via calcium release-activated calcium (CRAC)-mediated capacitive Ca(2+) entry, and stromal interaction molecule 1 (STIM1)- and Orai1-deficient cells are resistant to oxidant stress. Functionally, oxidant-induced Ca(2+) entry alters mitochondrial Ca(2+) handling and bioenergetics and triggers cell death. Sodium selenite is a salt, a colourless solid, and the most common water-soluble selenium compound. Sodium selenite induces apoptosis by ROS (reactive oxygen species)-mediated ER (endoplasmic reticulum) stress, activation of proapoptotic proteins Bad and Bax, Deltapsim disruption, release of cytochrome c, and consequent initiation of the pro-apoptotic “executioner” caspase cascade.
In normal cells, the levels of Ca(2+) are low in the mitochondria, whereas in apoptotic cells, Ca(2+) increases. Mitochondria uptake Ca(2+) via an inner membrane channel called the uniporter and extrude it into the cytoplasm through a Na(+)/Ca(2+) exchanger. Study shows that overload of Ca(2+) in the mitochondria in benzothiazepin (calcium channel blockers)-treated cells leads to its damage, thus affecting cellular function and survival. Calcium channel blockers did not induce apoptosis. However, combination of Calcium channel blockers and TRAIL (tumor necrosis factor-related apoptosis-inducing ligand) increased the apoptotic response approximately 25-fold compared with control. Increase in apoptosis followed enhanced levels of [Ca(2+)](m) and was accompanied by pronounced mitochondrial changes characteristic of mitochondria-mediated apoptosis. TRAIL-induced apoptosis is largely correlated with the expression levels of TRAIL death receptors on the cell surface.
Finally, Ca(2+) overload and Ca(2+)-dependent processes have been shown to activate and modulate the execution of a non-apoptotic death programme.
Mitochondria: a hub of redox activities and cellular distress control.
Mitochondrial oxidative stress: implications for cell death.
Reactive oxygen species in mitochondria-mediated cell death.
S-glutathionylation activates STIM1 and alters mitochondrial homeostasis.
Therapeutic advantage of combining calcium channel blockers and TRAIL in prostate cancer.
Specific mitochondrial calcium overload induces mitochondrial fission in prostate cancer cells.
A ceramide is a lipid molecule composed of the amino acid sphingosine and a fatty acid. Ceramides exist in great concentrations in the plasma membrane of a cell and act as signaling molecules for a number of cellular functions. Ceramide also have a role in certain pathological states, including cancer and inflammation. As an important lipid second messenger that mediates diverse signaling pathways, ceramide triggers cell functions including cell growth arrest, cell senescence, differentiation, proliferation, adhesion, cell migration, and apoptosis. It is believed to aid in suppressing the spread of cancer through some of these functions.
Study shows that ceramide-induced maturation is associated with the release of intracellular calcium stores. Ceramide caused a dose-dependent elevation in the second messenger IP(3). Elevation of IP(3), in turn, activated the IP(3) receptor calcium release channel on the ER (endoplasmic reticulum), resulting in a rise in cytoplasmic calcium. Thus study demonstrates that cross talk between the ceramide and phosphoinositide signaling pathways modulates intracellular calcium homeostasis.
Recently, carbonic anhydrase (CA) inhibitors have been proposed as a potential new class of antitumor agents. carbonic anhydrase (CA) inhibitors. Carbonic anhydrase (CA) inhibition can decrease cell proliferation and induce apoptosis in human tumor cells. The ability of carbonic anhydrase (CA) inhibitors to decrease pH(i) might trigger cell apoptosis through mediation of ceramide synthesis.
Acetazolamide (trade name Diamox) is used to treat glaucoma, epileptic seizures, benign intracranial hypertension (pseudotumor cerebri), altitude sickness, cystinuria, and dural ectasia. Acetazolamide is a carbonic anhydrase inhibitor, which means that it forces the kidneys to excrete bicarbonate (HCO3-), thus re-acidifying the blood. Carbonic acid inhibitors, such as acetazolamide, inhibit carbonic anhydrase (CA) in tissue and fluid, causing less movement of carbonic acid toward CO2 production. In the kidneys, blocking CA leads to bicarbonate wasting in the tubules (alkalizes urine), loss of bicarbonate subsequently leads to a metabolic acidosis. In the meantime, H+ backs up due to acetazolamide CA inhibition in the tubule and enter the cell with Cl-, then passes into the bloodstream, creating a hyperchloremic metabolic acidosis. This effect can also be used for therapeutic correction of respiratory alkalosis.
Carbonic anhydrase isoform IX (CA IX) is highly overexpressed in many types of cancer. Its expression, which is regulated by the HIF-1 transcription factor, is strongly induced by hypoxia and correlates with a poor response to classical chemo- and radiotherapies. CA contributes to acidification of the tumor environment by efficiently catalyzing the hydration of carbon dioxide to bicarbonate and protons, thereby leading to acquisition of metastatic phenotypes and chemoresistance to weakly basic anticancer drugs. Inhibition of this enzymatic activity by specific inhibitors, such as the sulfonamide indisulam, reverts these processes, establishing a clear-cut role for CA in tumorigenesis. Thus, selective CA inhibitors could prove useful for elucidating the role of CA in hypoxic cancers, for controlling the pH imbalance in tumor cells and for developing diagnostic or therapeutic applications for tumor management.
Ceramide triggers intracellular calcium release via the IP(3) receptor in Xenopus laevis oocytes.
Targeting tumor-associated carbonic anhydrase IX in cancer therapy.
In vivo targeting of tumor-associated carbonic anhydrases using acetazolamide derivatives.
Inhibition of carbonic anhydrase IX: a new strategy against cancer.
Recent studies show that Melatonin has significant anti-apoptotic effects. Melatonin inhibits the activation of mitochondrial death pathway such as upregulation of the expression of Bax and Bak, and the downregulation of Bcl-2 and BclxL. Melatonin also prevents mitochondrial translocation of Bax and prevents the collapse of mitochondrial membrane potential. Moreover, melatonin reduces activation of caspase-9 and caspase-3 induced by increases in Ca(2+) by blocking the release of cytochrome c and finally rescues cells from apoptosis. When you start cancer treatment, do not take Melatonin.



{kind=link}